Aging Processes in Organic Aerosols
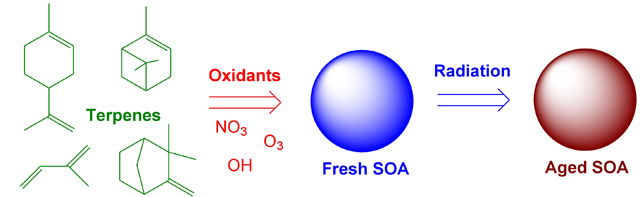
Chemical processes that convert volatile organic compounds (VOCs) into secondary organic aerosols (SOA) are reasonably fast. For example, terpenes rarely last longer than a few hours after they are released by vegetation on a hot sunny day. Once the condensable products of VOC oxidation partition into aerosol particles, the chemistry slows down considerably. However, it is incorrect to view SOA particles as a static collection of organic molecules which undergoes no chemistry after the initial particle condensation. A number of interesting aging processes leading to changes in the chemical composition and physical properties of aerosols occur on time scales ranging from minutes to days. Our group investigates mechanisms of photochemical and thermal (dark) aging in organic aerosols using a combination of spectroscopic and mass spectrometric techniques.
Aging of Organic Aerosols by Condensed-Phase Photochemistry
Direct photochemical aging of OA refers to processes initiated by absorption of solar radiation within the aerosol particles. This aging mechanism can be effective only if the following conditions are satisfied: (1) the organic aerosol material must have significant absorption in the tropospheric actinic window; (2) the yields for condensed-phase photochemical reactions, such as photodissociation, must be large compared to that for fluorescence, vibrational relaxation, geminate recombination, and various other non-reactive processes. We have found that SOA formed by ozonolysis and photooxidation of VOCs do indeed absorb light at atmospherically relevant wavelengths [107], leading to rich photochemistry within the particles such as direct photodissociation of peroxide [50] and carbonyl [57, 60] functional groups.
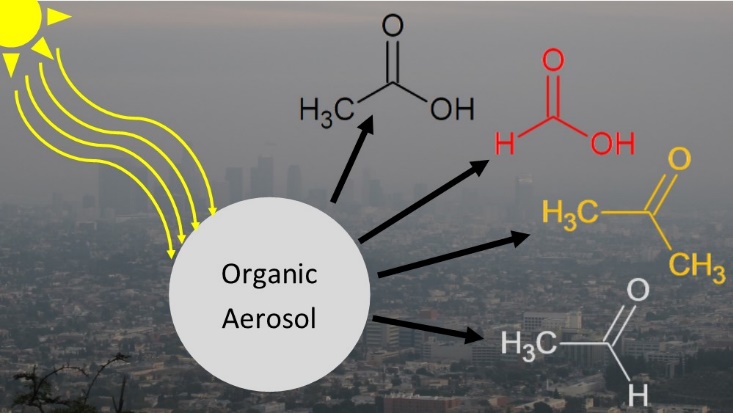 We have demonstrated the importance of condensed phase photochemistry in aging of organic aerosols. For example, we found that condensed-phase photochemical processes in alpha-pinene SOA result in a decrease in mass and diameter of particles occurring on atmospherically-relevant time scales [98]. Photodegradation of SOA produces a variety of small, oxygenated VOCs, which evaporate from the particles leading to the observed mass loss. This photodegradation process could produce upwards of a few Tg/year of formic acid in the atmosphere comparable to its primary sources [111]. However, uptake of VOCs during irradiation is also possible in a so-called photosensitized process. We showed that photosensitized uptake of limonene on SOA particles occurs in competition with the photodegradation, but that the photodegradation occurs on a much faster time scale, and is therefore more important aerosol aging [122]. We have demonstrated the importance of condensed phase photochemistry in aging of organic aerosols. For example, we found that condensed-phase photochemical processes in alpha-pinene SOA result in a decrease in mass and diameter of particles occurring on atmospherically-relevant time scales [98]. Photodegradation of SOA produces a variety of small, oxygenated VOCs, which evaporate from the particles leading to the observed mass loss. This photodegradation process could produce upwards of a few Tg/year of formic acid in the atmosphere comparable to its primary sources [111]. However, uptake of VOCs during irradiation is also possible in a so-called photosensitized process. We showed that photosensitized uptake of limonene on SOA particles occurs in competition with the photodegradation, but that the photodegradation occurs on a much faster time scale, and is therefore more important aerosol aging [122].
We quantified photodegradation rates of SOA by condensed-phase photochemical processes over many days of UV exposure [158]. The experiments relied on a quartz crystal microbalance to quantify the mass loss rate from various SOA materials. The initial mass loss rate was high (corresponding to 1-5 % fractional mass loss per hour) but it slowed down after 24 h of irradiation with a photostable fraction of SOA degrading much slower. These experiments have confirmed that condensed-phase photochemistry is an important aging mechanism for SOA during long-range transport, but hard to parametrize in climate models. In a follow-up study, we discovered that this long-term exposure of SOA to ultraviolet radiation leads to a remarkable increase in viscosity by as much as five orders of magnitude [173]. This UV exposure can lead to an increased abundance of aerosols in the glassy solid state in the troposphere, with important implications for climate predictions, since aerosols in the glassy solid state can act as nuclei for ice clouds thereby influencing cloud radiative and microphysical properties. Since the UV radiation causes the mixing time within SOA to be extremely long for most of the troposphere, and considerably longer than assumptions frequently used in chemical transport models, current chemical transport models may have incorrectly predicted the growth, evaporation, and size of SOA particles, and ultimately air quality and climate. Overall, our results clearly demonstrated that UV exposure needs to be considered when predicting the environmental impacts of SOA.
Aqueous Photochemistry of Orgainic Aerosols
 An important avenue of our research is photochemistry of SOA in aqueous environments, which is relevant for understanding cloud-processing of organic aerosols. In our first paper on this topic, we reported the effect of UV irradiation on the molecular composition of aqueous extracts of limonene ozonolysis SOA. We showed that photolysis had a significant effect on the composition of the dissolved organics: oligomeric compounds were destroyed, carbonyl compounds were photolyzed, carboxylic acids were generated during photolysis, and large organic peroxides were recycled into smaller peroxides. In a related study on high-NOx isoprene SOA [80] we discovered new photochemical reactions converting organic nitrates into unusual heterocyclic compounds containing nitrogen. In our follow up studies, we showed that aqueous photodegradation is common to a broad range of SOA [93, 100, 118]. We found that the SOA material became more volatile on average after the photolysis [118]. These results suggest that SOA dissolved in cloud/fog droplets should undergo significant photolytic processing on a time scale of hours to days, and may need to be considered by atmospheric models. We have also examined the role of co-dissolved salts on photochemistry of SOA, from relatively small concentrations characteristic of fog droplets [153] to molar concentrations characteristic of aerosol liquid water [174]. We are continuing these experiments in collaboration with the group of Prof. Annmarie Carlton. An important avenue of our research is photochemistry of SOA in aqueous environments, which is relevant for understanding cloud-processing of organic aerosols. In our first paper on this topic, we reported the effect of UV irradiation on the molecular composition of aqueous extracts of limonene ozonolysis SOA. We showed that photolysis had a significant effect on the composition of the dissolved organics: oligomeric compounds were destroyed, carbonyl compounds were photolyzed, carboxylic acids were generated during photolysis, and large organic peroxides were recycled into smaller peroxides. In a related study on high-NOx isoprene SOA [80] we discovered new photochemical reactions converting organic nitrates into unusual heterocyclic compounds containing nitrogen. In our follow up studies, we showed that aqueous photodegradation is common to a broad range of SOA [93, 100, 118]. We found that the SOA material became more volatile on average after the photolysis [118]. These results suggest that SOA dissolved in cloud/fog droplets should undergo significant photolytic processing on a time scale of hours to days, and may need to be considered by atmospheric models. We have also examined the role of co-dissolved salts on photochemistry of SOA, from relatively small concentrations characteristic of fog droplets [153] to molar concentrations characteristic of aerosol liquid water [174]. We are continuing these experiments in collaboration with the group of Prof. Annmarie Carlton.
The Role of Molecular Environment in Photochemistry of Particulate Organics
Our group has performed experiments on photochemistry of SOA material in a variety of matrices, where we control the viscosity of SOA (by adjusting temperature and humidity), and/or the embedded molecules. To investigate the role of the particle viscosity on photochemistry, we investigated photolysis of various molecules embedded in alpha-pinene SOA at high (liquid) and low (glassy) temperatures and showed that photochemistry is suppressed in the glassy state [91]. In a follow up study, we examined this effect in three types of SOA and found that the photochemistry is suppressed at lower temperature or lower relative humidity in all three model systems [110]. We investigated the photolysis of 4-nitrocatechol and 2,4-dinitrophenol in semisolid isomalt as a new type of surrogate for glassy SOA and compared it to photolysis in liquid water, isopropanol, and octanol showing remarkable sensitivity of photochemistry of these compounds to the surrounding matrix [168]. We also studied photooxidation of toluene, the brown color of which is mostly due to nitrophenols, and found that toluene SOA lifetime with respect to photobleaching and lifetimes of individual chromophores in SOA with respect to photodegradation depend strongly on the sample matrix in which SOA compounds are exposed to sunlight [174].
Photochemistry of Selected Atmospheric Compounds
In an effort to better understand mechanisms of atmospheric photochemical processes, we conduct fundamental experimental and computational studies of photodissociation of important atmospheric molecules in gaseous phase and condensed phases. For example, we found that photolysis of the simplest organic peroxide CH3OOH embedded in ice clusters results in rich photochemistry on a picosecond time scale [63]. This was followed by a combined experimental and theoretical investigation of photodissociation quantum yields and absorption cross sections of CH3OOH in water and in ice [78]. Another study involved the absorption spectra and aqueous photochemistry of alkyl nitrates [104]. We determined that the aqueous photolysis of the alkyl nitrates was insignificant compared to the gas-phase photodegradation and aqueous OH reactions. We have investigated the mechanisms of photochemistry of cyclohexanone, an atmospherically-relevant ketone [115], cis-pinonic acid, a major product of oxidation of alpha-pinene [90], as well photochemistry of aldehydes [97].
More recently, we examined photochemistry of nitrophenols, important air pollutants that contribute to the brownish color of particulate matter. We investigated the photolysis of 4-nitrocatechol (4NC) and 2,4-dinitrophenol (24DNP) in semisolid isomalt as a new type of surrogate for glassy SOA and compared it to photolysis in liquid water, isopropanol, and octanol [168]. The quantum yield of 4NC photolysis was smaller in an isomalt glass than in isopropanol. Both 4NC and 24DNP had much lower photolysis rates in water than in organic matrices. In isopropanol solution, most products appeared to result from the oxidation of 4NC, in stark contrast to photoreduction and dimerization products that were observed in solid isomalt. Therefore, the photochemical fate of 4NC, and other nitrophenols, should depend on whether they undergo photodegradation in a liquid or semisolid organic particle. We examined the influence of solvent on the electronic structure and photochemistry of nitrophenols by comparing the photodegradation of four nitrophenols in organic solvent and aqueous solution [183]. We found the solvent effects on the electronic structure and photodegradation processes were minimal between 2-propanol and water for most of the nitrophenols. One of the nitrophenols studied, 2,4-dinitrophenol, exhibited dramatically increased reactivity in organic solvent compared to that in water, suggesting that it will have short lifetimes in atmospheric organic aerosol particles. To better understand the mechanism of photodegradation of nitrophenols in both aqueous and organic solvents, we did an experimental study of ultrafast electron dynamics in 4-nitrocatechol, a common product of biomass burning and fossil fuel combustion [191]. Contrary to our initial expectations, we found that electronic triplet states are not efficiently populated upon 340 nm excitation, as efficient proton transfer occurs in the excited state on a time scale of a few picoseconds in water and tens of picoseconds in 2-propanol. This suggests that triplet states do not play a significant role in the photochemical reactions of 4-nitrocatechol in the environment and, by extension, in nitrophenols in general. This is an important deviation from the currently accepted wisdom, which attributes excited state chemistry in environmental organics to triplet states rather than photoacids.
Photochemical Formation of Reactive Oxygen Species (ROS)
We recently started an exciting collaboration with the group of Prof. Manabu Shiraiwa to study mechanisms of ROS formation in aerosol and cloud droplets irradiated by sunlight, using electron paramagnetic resonance (EPR) spectroscopy with a spin-trapping agent BMPO and high-resolution mass spectrometry. Establishing the mechanism of ionization of BMPO-radical adducts was a key to this project [199]. We examined the photosensitized formation of ROS in mixtures of benzoquinone and levoglucosan, common components of biomass smoke [188]. The EPR analysis showed that the irradiation of these mixtures leads to the formation of OH, H, and organic radicals (R/RO), which can perpetuate radical chain mechanisms within aqueous droplets. The mass spectrometric analysis provided a way to distinguish BMPO adducts containing benzoquinone and levoglucosan derivatives, confirming that aqueous photochemistry can transform innocuous chemicals, like levoglucosan, into organic radicals that lead to increasingly complex chemistry within the droplets. The results of a kinetic model showed that OH rapidly reacts with levoglucosan and can shorten its atmospheric lifetime, which has important implications for its application as a tracer molecule in field studies. These results imply that photoirradiation of aerosols containing photosensitizers induces ROS formation and secondary radical chemistry to drive photochemical aging of BBOA in the atmosphere.
We combined an in situ UVvis irradiation system with EPR spectroscopy to characterize the photolytic formation of ROS in aqueous extracts of SOA formed by the oxidation of isoprene, alpha-pinene, alpha-terpineol, and toluene [192]. We observed substantial formation of free radicals, including OH, superoxide (HO2), and organic radicals (R/RO) upon irradiation. The types of detected radicals and aqueous photolysis of model compounds indicated that photolysis of carbonyls by Norrish type I mechanisms plays an important role in the organic radical formation. This photolytic ROS formation serves as the driving force for cloud and fog processing of SOA.
Role of Photosensitizers in Aging of Aerosols
In collaboration with the group of Dr. Kristopher McNeill and Dr. Nadine Borduas-Dedekind, we evaluated the ability of SOA compounds to produce singlet oxygen, 1O2 [149]. We found that SOA from aromatic precursors are able to efficiently sensitize 1O2. We predicted that certain organic compounds found in aerosols, such as amino acids, have shortened lifetimes by more than a factor of two when 1O2 is considered as an additional sink. As a spin-off of this study, we also examined the effect of SOA aqueous photochemistry on its ice nucleation ability [155]. We found that UV-B exposure of SOA increased the particle hygroscopicity and thus its ability to from cloud droplets.
In collaboration with the group of Dr. Christian George at IRCELYON, we examined chemical reactions of triplet-excited states of SOA molecules. We reported that photosensitized chemistry can be an efficient mechanism of sulfate oxidation, and potentially responsible for unexplained rapid growth of particulate matter in megacities, such as Shanghai [152]. We examined the photosensitized chemistry of three aromatic ketones (xanthone, flavone, and acetophenone) and also of SOA arising from the photooxidation of naphthalene by means of transient absorption spectroscopy [162]. We found that deoxygenated naphthalene SOA solutions had a strong transient absorption after a pulsed UV excitation, and were also quenched by iodide ions similar to the single ketone experiments indicating that compounds in naphthalene SOA can act as photosensitizers. We investigated the photosensitizing properties of SOA formed during the OH-initiated oxidation of naphthalene [166]. A remarkable result of that work was observation of a photosensitized production of sulfate in the presence of SO2. Dark formation of sulfate on naphthalene SOA was also observed. As naphthalene and other polycyclic aromatics are important SOA precursors in the urban and suburban areas, these dark and photosensitized reactions are likely to play an important role in sulfate and SOA formation.
Thermal and Acid-Catalyzed Aging of Organic Aerosols
In addition to the photochemical aging experiments, we also investigate chemical changes in SOA resulting from thermal reactions. We explored the effects of water on the chemical composition of SOA during long-term aging processes [167]. We found that chemical composition of SOA did change during long-term (several days) exposure to water vapor and liquid water, but the extent of the change was surprisingly small. We reported experiments on hydrolysis of oxaloacetic acid (OAA) catalyzed by ammonium ion [165]. This catalysis has not been studied under atmospherically relevant conditions despite interest in OAA in the atmosphere. We found that under moderately acidic conditions representative of aerosol particles (pH=3-4), the decarboxylation rate of OAA increased linearly with ammonium concentration, with lifetime becoming as short as one hour under typical aerosol liquid water conditions. This result explains why OAA is hard to detect in field measurements even though it is a known oxidation product of succinic acid.
We worked with the Shiraiwa group members to better understand formation of ROS during dissolution of SOA in water. We found formation of superoxide (O2) with molar yields of 0.010.03% from aqueous reactions of biogenic SOA generated by low-NOx photooxidation of isoprene, beta-pinene, alpha-terpineol, and d-limonene [160]. We later extended these results to SOA produced under high-NOx conditions, characteristic of urban environments [180]. Furthermore, we determined that biomass burning and pyrolysis aerosol particles contain persistent free radicals and produce reactive oxygen species when dissolved in water [177]. These findings help better understand oxidative stress upon respiratory deposition of SOA particles in lungs.
We examined the composition and optical properties of SOA exposed to the highly acidic conditions [181], and showed that acidity can be a major driver of SOA aging in areas with high concentrations of H2SO4, such as in the upper troposphere and lower stratosphere. To better understand the mechanism of this acid-catalyzed process, we teamed up with the group of Prof. Scott Rychnovsky to help identify the chromophores formed by acid-catalyzed transformations of common alpha-pinene oxidation products, namely, cis-pinonic acid and cis-pinonaldehyde [193]. The latter compounds was proven to drive the mechanism of browning of the aged SOA by forming conjugated products, which we could identify, by an acid-catalyzed aldol condensation. This chemistry could be relevant for environments characterized by high sulfuric acid concentrations, for example, during the transport of organic compounds from the lower to the upper atmosphere by fast updrafts.
|

